pVACtools¶
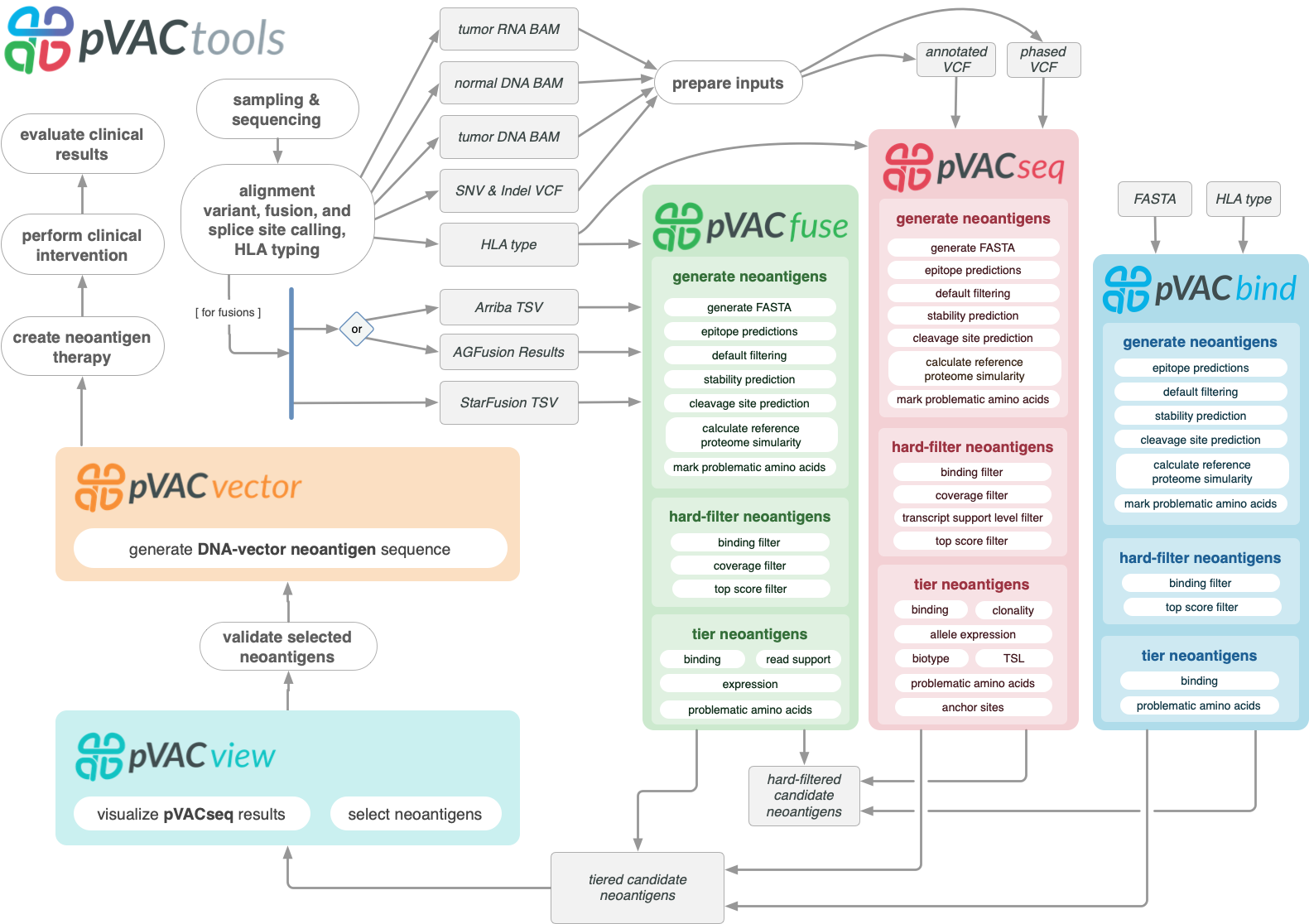
pVACtools is a cancer immunotherapy tools suite consisting of the following tools:
- pVACseq
A cancer immunotherapy pipeline for identifying and prioritizing neoantigens from a VCF file.
- pVACbind
A cancer immunotherapy pipeline for identifying and prioritizing neoantigens from a FASTA file.
- pVACfuse
A tool for detecting neoantigens resulting from gene fusions.
- pVACvector
A tool designed to aid specifically in the construction of DNA-based cancer vaccines.
- pVACview
An application based on R Shiny that assists users in reviewing, exploring and prioritizing neoantigens from the results of pVACtools processes for personalized cancer vaccine design.
Contents¶
New in Version 3.1¶
This version adds the following features, outlined below. Please note that pVACtools 4.0 is not backwards-compatible and certain changes will break old workflows.
Breaking Changes¶
pVACseq|pVACfuse|pVACbind report files have been reformatted to add some additional information and, in the case of pVACfuse and pVACbind, remove columns where all values were
NA. Existing output files will no longer work with the standalone commands as well as pVACview.The format of the Mutation Position column has been updated to no longer use 0 and n+1 to denote mutations starting before or ending after the epitope. This column now only shows the actually mutated positions.
New Features¶
We now support MHCflurry and NetMHCpanEL elution algorithms.
Users are now able to select specific amino acids that would be problematic for vaccine manufacturing and have the pipelines mark epitopes with such amino acids.
When running the reference proteome similarity step, users are now able to specify a peptide fasta to search against instead of using BLAST. Any exact matches against the entries in the peptide fasta are counted as a hit.
The aggregate report now takes into account many command line thresholds when tiering candidates. We also refined the way we determine the Best Peptide to take into account the biotype and TSL of the transcripts coding for the peptide, and whether or not the candidate has any problematic positions or fails the anchor criteria. Please see the output file section of the documentation for more details.
pVACview has been updated with a host of new features
Users may adjust a wider variety of thresholds for retiering.
Users are now able to reset the tiering thresholds to the ones originally used when running pVACview.
Transcripts resulting in the same set of epitope candidates are now grouped together to make it easier to identify unique candidates.
Elution data is displayed in the epitope details section of pVACview.
Reference match details are displayed in the transcript set details section of pVACview.
pVACfuse now supports output files from Arriba for fusion peptide predictions.
Users may provide an optional STAR-fusion output file to their pVACfuse run in order to extract expression and read support data for their candidates. These will be used for filtering, as well as for tiering in the aggregate report. Please see the output file section of the documention for more details.
When running the
pvacseq generate_protein_fastacommand, users are now able to specify an aggregated report as the--input-tsv. When using such a TSV, they can also use the--aggregate-report-evaluationto specify Evaluation statuses to include in the protein fasta. This is useful when creating a peptide fasta for vaccine ordering after using pVACview to select vaccine candidates and exporting the results to a new TSV.
Minor Changes¶
The reference proteome step is now run on the aggregated report instead of the filtered report.
A new parameter
--aggregate-inclusion-binding-thresholdcontrols which epitope candidates are included in the aggregate report.
Past release notes can be found on our Release Notes page.
To stay up-to-date on the latest pVACtools releases please join our Mailing List.
Citations¶
Jasreet Hundal , Susanna Kiwala , Joshua McMichael, Chris Miller, Huiming Xia, Alex Wollam, Conner Liu, Sidi Zhao, Yang-Yang Feng, Aaron Graubert, Amber Wollam, Jonas Neichin, Megan Neveau, Jason Walker, William Gillanders, Elaine Mardis, Obi Griffith, Malachi Griffith. pVACtools: A Computational Toolkit to Identify and Visualize Cancer Neoantigens. Cancer Immunology Research. 2020 Mar;8(3):409-420. doi: 10.1158/2326-6066.CIR-19-0401. PMID: 31907209.
Jasreet Hundal, Susanna Kiwala, Yang-Yang Feng, Connor J. Liu, Ramaswamy Govindan, William C. Chapman, Ravindra Uppaluri, S. Joshua Swamidass, Obi L. Griffith, Elaine R. Mardis, and Malachi Griffith. Accounting for proximal variants improves neoantigen prediction. Nature Genetics. 2018, DOI: 10.1038/s41588-018-0283-9. PMID: 30510237.
Jasreet Hundal, Beatriz M. Carreno, Allegra A. Petti, Gerald P. Linette, Obi L. Griffith, Elaine R. Mardis, and Malachi Griffith. pVACseq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Medicine. 2016, 8:11, DOI: 10.1186/s13073-016-0264-5. PMID: 26825632.
Source code¶
The pVACtools source code is available in GitHub.
License¶
This project is licensed under BSD 3-Clause Clear License.