Output Files¶
The pVACseq pipeline will write its results in separate folders depending on which prediction algorithms were chosen:
MHC_Class_I: for MHC class I prediction algorithmsMHC_Class_II: for MHC class II prediction algorithmscombined: If both MHC class I and MHC class II prediction algorithms were run, this folder combines the neoepitope predictions from both
Each folder will contain the same list of output files (listed in the order created):
File Name |
Description |
|---|---|
|
An intermediate file with variant, transcript, coverage, vaf, and expression information parsed from the input files. |
|
The above file but split into smaller chunks for easier processing with IEDB. |
|
A fasta file with mutant and wildtype peptide subsequences for all processable variant-transcript combinations. |
|
A list of all predicted epitopes and their binding affinity scores, with
additional variant information from the |
|
The above file after applying all filters, with (optionally) cleavage site, stability predictions, and reference proteome similarity metrics added. |
|
A file outlining details of reference proteome matches |
|
An aggregated version of the |
Filters applied to the filtered.tsv file¶
The filtered.tsv file is the all_epitopes file with the following filters applied (in order):
Binding Filter
Coverage Filter
Transcript Support Level Filter
Top Score Filter
Please see the Standalone Filter Commands documentation for more information on each individual filter. The standalone filter commands may be useful to reproduce the filtering or to chose different filtering thresholds.
all_epitopes.tsv and filtered.tsv Report Columns¶
Column Name |
Description |
|---|---|
|
The chromosome of this variant |
|
The start position of this variant in the zero-based, half-open coordinate system |
|
The stop position of this variant in the zero-based, half-open coordinate system |
|
The reference allele |
|
The alt allele |
|
The Ensembl ID of the affected transcript |
|
The transcript support level (TSL)
of the affected transcript. |
|
The Ensembl ID of the affected gene |
|
The type of variant. |
|
The amnio acid change of this mutation |
|
The protein position of the mutation |
|
The Ensembl gene name of the affected gene |
|
The HGVS coding sequence variant name |
|
The HGVS protein sequence variant name |
|
The HLA allele for this prediction |
|
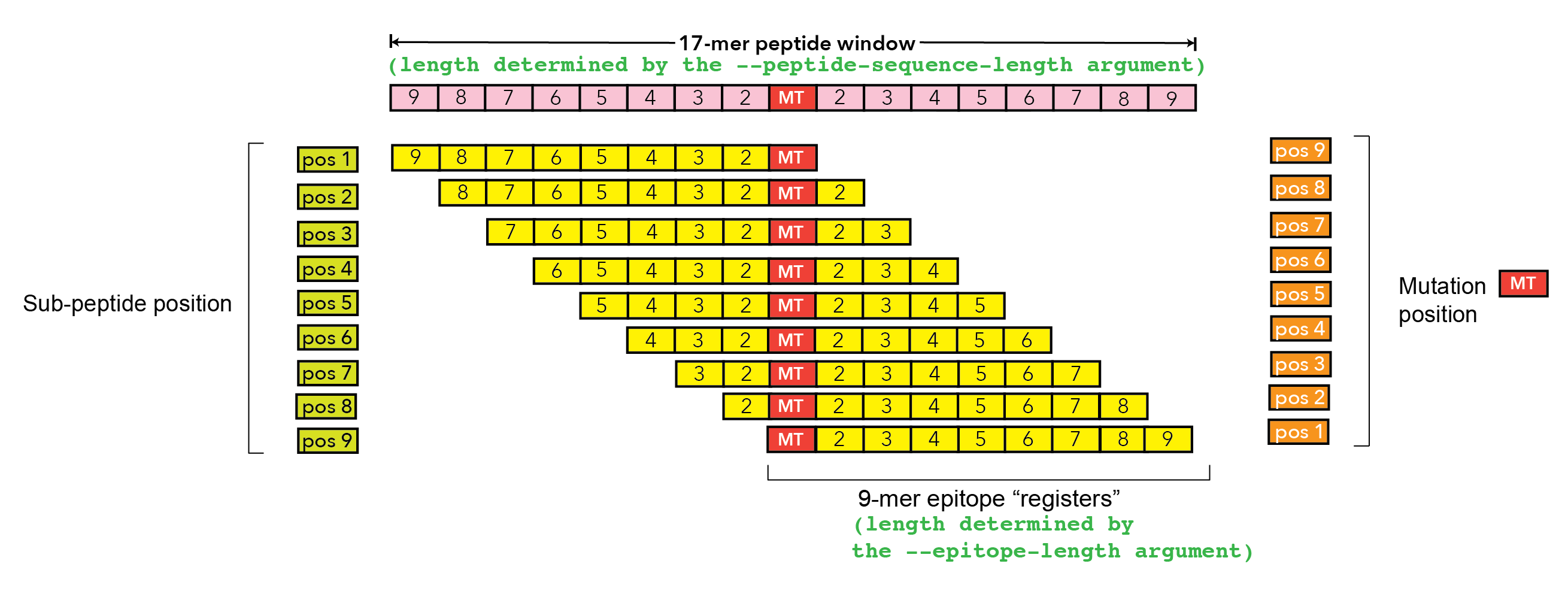
The peptide length of the epitope |
|
The one-based position of the epitope within the protein sequence used to make the prediction |
|
The one-based position of the start of the mutation within the epitope sequence. |
|
The mutant epitope sequence |
|
The wildtype (reference) epitope sequence at the same position in the full protein sequence. |
|
Prediction algorithm with the lowest mutant ic50 binding affinity for this epitope |
|
Lowest ic50 binding affinity of all prediction algorithms used |
|
ic50 binding affinity of the wildtype epitope. |
|
|
|
Prediction algorithm with the lowest binding affinity percentile rank for this epitope |
|
Lowest percentile rank of this epitope’s ic50 binding affinity of all prediction algorithms used (those that provide percentile output) |
|
binding affinity percentile rank of the wildtype epitope. |
|
Tumor DNA depth at this position. |
|
Tumor DNA variant allele frequency (VAF) at this position. |
|
Tumor RNA depth at this position. |
|
Tumor RNA variant allele frequency (VAF) at this position. |
|
Normal DNA depth at this position. |
|
Normal DNA variant allele frequency (VAF) at this position. |
|
Gene expression value for the annotated gene containing the variant. |
|
Transcript expression value for the annotated transcript containing the variant. |
|
Median ic50 binding affinity of the mutant epitope across all prediction algorithms used |
|
Median ic50 binding affinity of the wildtype epitope across all prediction algorithms used.
|
|
|
|
Median binding affinity percentile rank of the mutant epitope across all prediction algorithms (those that provide percentile output) |
|
Median binding affinity percentile rank of the wildtype epitope across all prediction algorithms used (those that provide percentile output)
|
|
ic50 binding affintity and percentile ranks for the |
|
A unique idenitifer for this variant-transcript combination |
|
Mean hydropathy of last 7 residues on the C-terminus of the peptide |
|
Max GRAVY score of any kmer in the amino acid sequence. Used to determine if there are any extremely hydrophobic regions within a longer amino acid sequence. |
|
Is N-terminal amino acid a Glutamine, Glutamic acid, or Cysteine? |
|
Is the C-terminal amino acid a Cysteine? |
|
Is the C-terminal amino acid a Proline? |
|
Number of Cysteines in the amino acid sequence. Problematic because they can form disulfide bonds across distant parts of the peptide |
|
Is the N-terminal amino acid a Asparagine? |
|
Number of Asparagine-Proline bonds. Problematic because they can spontaneously cleave the peptide |
|
Position of the highest predicted cleavage score |
|
Highest predicted cleavage score |
|
List of all cleavage positions and their cleavage score |
|
Stability of the pMHC-I complex |
|
Half-life of the pMHC-I complex |
|
The % rank stability of the pMHC-I complex |
|
Nearest neighbor to the |
|
Was there a BLAST match of the mutated peptide sequence to the reference proteome? |
filtered.tsv.reference_matches Report Columns¶
This file is only generated when the --run-reference-proteome-similarity
option is chosen.
Column Name |
Description |
|---|---|
|
The chromosome of this variant |
|
The start position of this variant in the zero-based, half-open coordinate system |
|
The stop position of this variant in the zero-based, half-open coordinate system |
|
The reference allele |
|
The alt allele |
|
The Ensembl ID of the affected transcript |
|
The peptide sequence submitted to BLAST |
|
The BLAST alignment hit ID (reference proteome sequence ID) |
|
The BLAST alignment hit definition (reference proteome sequence name) |
|
The BLAST query sequence |
|
The BLAST match sequence |
|
The match start position in the matched reference proteome sequence |
|
The match stop position in the matched reference proteome sequence |
all_epitopes.aggregated.tsv Report Columns¶
The all_epitopes.aggregated.tsv file is an aggregated version of the all_epitopes TSV.
It presents the best-scoring (lowest binding affinity)
epitope for each variant, and outputs additional binding affinity, expression, and
coverage information for that epitope. It also gives information about the
total number of well-scoring epitopes for each variant, the number of
transcripts covered by those epitopes, as well as the HLA alleles that those
epitopes are well-binding to. Lastly, the report will bin variants into tiers
that offer suggestions as to the suitability of variants for use in vaccines.
Column Name |
Description |
|---|---|
|
For each HLA allele in the run, did the mutation result in an epitope that bound well to the HLA allele? (with median mutant binding affinity < 1000). |
|
The Ensembl gene name of the affected gene |
|
The amino acid change for the mutation |
|
The number of transcripts for this mutation that resulted in at least one well-binding peptide (median mutant binding affinity < 1000). |
|
The best-binding mutant epitope sequence (lowest median mutant binding affinity) |
|
The one-based position of the start of the mutation within the epitope sequence. |
|
The number of unique well-binding peptides for this mutation. |
|
Median ic50 binding affinity of the best-binding mutant epitope across all prediction algorithms used |
|
Median ic50 binding affinity of the corresponding wildtype epitope across all prediction algorithms used. |
|
Median binding affinity percentile rank of the best-binding mutant epitope across all prediction algorithms used (those that provide percentile output) |
|
Median binding affinity percentile rank of the corresponding wildtype epitope across all prediction algorithms used (those that provide percentile output) |
|
Gene expression value for the annotated gene containing the variant. |
|
Tumor RNA variant allele frequency (VAF) at this position. |
|
Tumor RNA depth at this position. |
|
Tumor DNA variant allele frequency (VAF) at this position. |
|
A tier suggesting the suitability of variants for use in vaccines. |
The pVACseq Aggregate Report Tiers¶
To bin a variant in a tier, the best binding epitope is evaluated as follows:
Tier |
Citeria |
|---|---|
|
Mutant allele is not expressed |
|
Mutant allele has low expression (TPM * RNA_VAF < 1) |
|
Likely not in the founding clone of the tumor (DNA_VAF > max(DNA_VAF)/2) |
|
Mutation is at an anchor residue in the shown peptide, and the WT allele has good binding (WT ic50 <1000) |
|
Fails two or more of the above criteria |
|
Passes the above criteria, has decent MT binding (ic50 < 1000) |
|
Passes the above criteria, has strong MT binding (ic50 < 1000) and strong expression (TPM * RNA_VAF > 3) |