Features¶
When visualizing and exploring your neoantigen candidates, pVACview starts by providing an overview of all variants used to predict neoantigen candidates. You can then investigate each variant in greater detail by breaking them down to the different transcripts that generate good-binding peptides. These transcripts then often each produce a different set of peptides that are predicted to bind well to certain HLA alleles. The individual MT/WT peptides can be further explored in terms of the distribution of binding algorithm predictions as well as anchor locations. Here below, we walk through the different sections of the pVACview explore page in detail.
Main aggregate report table¶
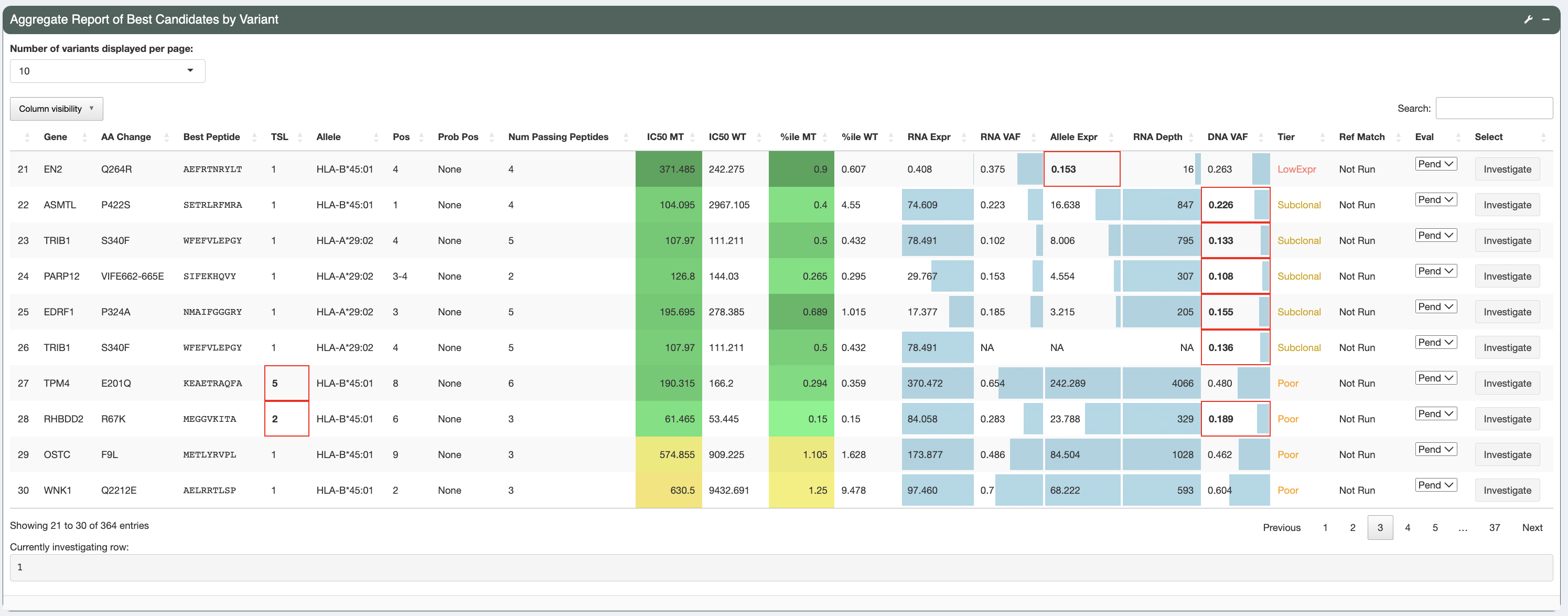
This is the aggregate report table generated as output from the pVACseq pipeline. The aggregate report summarizes the best-scoring epitope for each variant and outputs additional information specific to that epitope. It also provides information on a high level, showing the total number of well-scoring epitopes for each variants and transcript that cover them. There is a tiering giving to each variant based on a set of evaluation rules, helping you in prioritizing which variants to focus on first. Details on the report columns can be found here.
As shown, different cells of the report table are highlighted in various ways:
- IC50 MT and %ile MT columns:
The IC50 MT and %ile MT columns represent the median/lowest predicted IC50 binding affinities and median/lowest predicted binding percentiles. Whether these columns contain median or lowest data depends on the value set for the
--top-score-metricin the original pVACseq run. Different colors are used to give a general idea of where predictions fall in relation to the binding threshold and percentile threshold set:IC50 MT < binding threshold and %ile MT < percentile threshold colored with shades of green
binding threshold < IC50 MT < (2 * binding threshold) and percentile threshold < %ile MT < (2 * percentile threshold) colored with shades of orange
IC50 MT > (2 * binding threshold) and %ile MT > (2 * percentile threshold) colored with red
- RNA expression, RNA VAF, Allele Expr, RNA Depth, DNA VAF columns:
These columns have bar graphs as cell backgrounds to give an idea where specific values fall across the entire patient sample:
RNA VAF and DNA VAF columns range from 0 to 1
RNA Depth ranges from 0 to the max depth across all rows
RNA expression ranges from 0 to 50. This is done in order to highlight expression values that are in the lower range, based on the reasoning that, for neoantigen candidates, when the RNA expression reaches a certain level it is considered expressed without the need to specify how high the exact RNA expression value is.
Allele Expression ranges from 0 to max of RNA VAF column multiplied by 50 (which is the max for the RNA expression bar graph range).
The
Tiercolumn is colored based on the specific cell value and red boxes across the row are used to highlight values that did not pass filters and resulted in the tier given.
For more details on what each column represent, you can hover over the column names and/or click the tool icon on the top right where Help documentation is located.
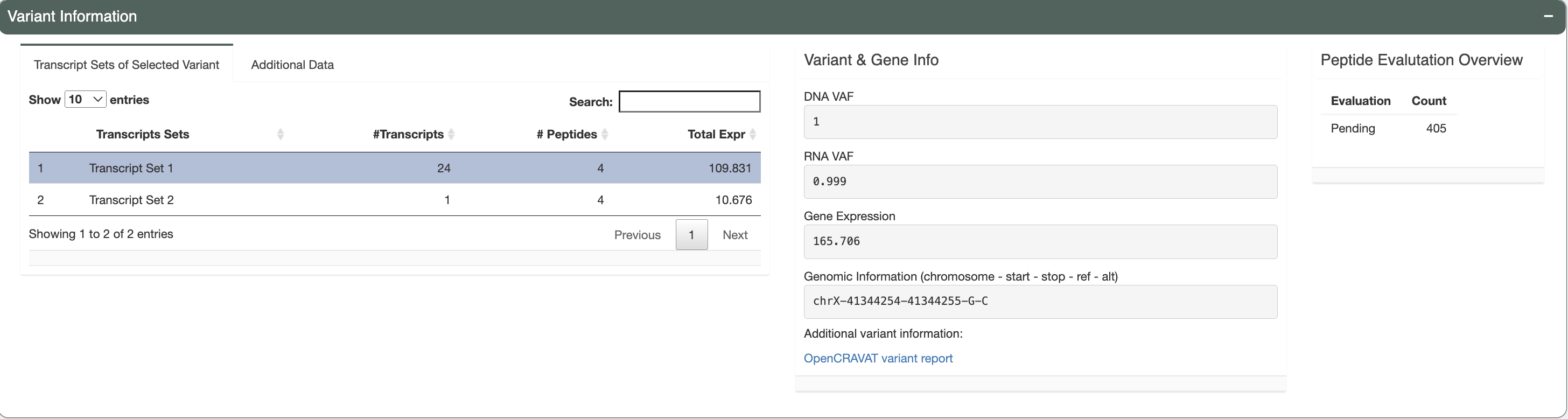
Variant Information (Transcripts, gene info and evaluation overview)¶
There are three separate boxes in this section as shown, from left to right you have:
Variant Information tabs
Transcript Sets of Selected Variant
The selected variant may have multiple transcripts covering the region. However, some transcripts might code for the same set of neoantigen candidates if the difference between transcripts is outside of the immediate area around the somatic mutation of interest. This table lists these different sets with the number of transcripts in each set, the number of corresponding peptides and the total expression of all transcripts in the set.
Referene Matches
When the reference proteome similarity feature was enabled during the original pVACseq run, this tab will show the Best Peptide, the larger peptide sequence around the Best Peptide that was queried for and it will list the hits that were found in the reference proteome for that query sequence.
Additional data
The data displayed in this tab is dependent on the additional data file that you provided in the
Uploadpage. The IC50 MT value and %ile MT values are shown if the app was able to locate the same variant in the data file provided. Values will show up as N/A if IC50 MT or %ile MT values are not provided in the additional file. Additionaly, the Best Peptide of the variant from that file will be listed as well as the HLA Allele the Best Peptide prediction was binding to and the Best Transcript for the prediction.
Variant & Gene Info
This box displays the DNA VAF, RNA VAF, and gene expression values for the variant you have selected for investigation. The genomic information is provided in the format showing the chromosomal location of the variant for further variant analysis such as manual review. We also provide a link out to the variant report provided by OpenCRAVAT. This report will allow users to explore the variant with information regarding: variant annotation, cancer, population allele frequencies, clinical relevance, gene annotation, pathogenicity prediction etc.
Peptide Evaluation Overview
This box shows an overview of the current state of your evaluation choices (i.e. how many are marked accepted, rejected, or for review). Note that aggregate reports generated from the pvacseq pipeline defaults everything to
Pending.
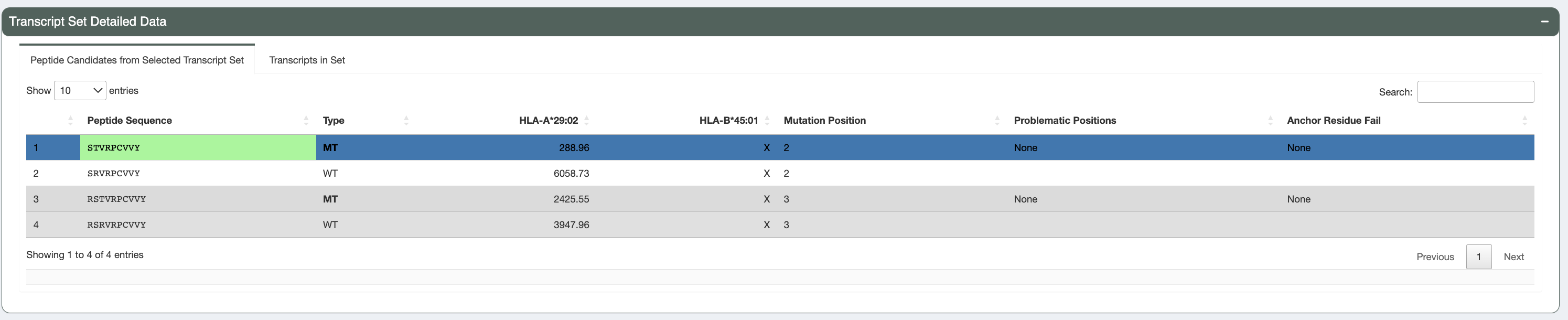
Transcript Set Detailed Data (Peptide Information)¶
Upon selecting the transcript set for further examination, users can navigate to this table which displays
all peptide sequences (from your selected transcript set in the transcripts table) that were predicted to be good binders
(for at least 1 HLA allele). Both mutant (MT) and wildtype (WT) sequences
are shown, along with binding affinities (where the MT binding passed the binding threshold). Whether this table shows
mutant or lowest binding affinities depends on the value of the
--top-score-metrics from the original pVACseq run.
This table also shows for each peptide, whether there were any problematic positions and whether or not the peptide failed the anchor residue criteria for any of the HLA alleles. Peptides failing these criteria are deprioritized in the sorting of this table.
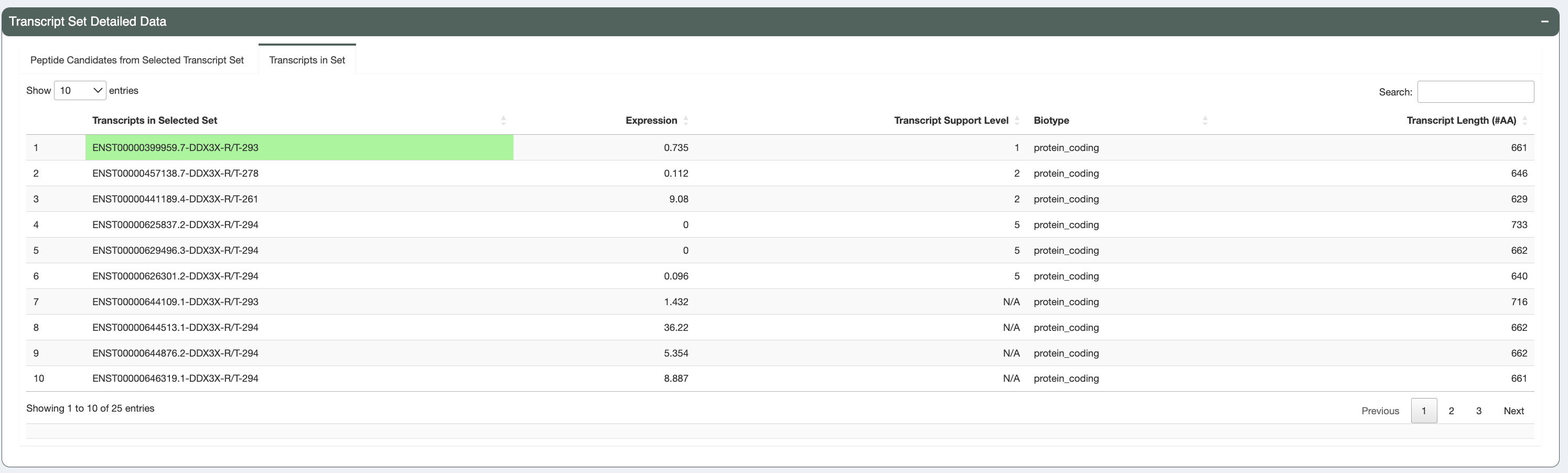
We also provide (in the “Transcripts in Set” tab), additional information regarding the transcripts producing these peptides. This includes: transcript id, individual transcript expression, transcript support level, biotype and transcript length. Transcripts with a protein_coding biotype, low TSL and long length are prioritized in the sorting of this table.
Additional Peptide Information (Additional information regarding individual algorithm binding and anchor scores)¶
There are five different tabs in this section of the app, providing peptide-level details on the MT/WT peptide pair that you selected in the peptide table.
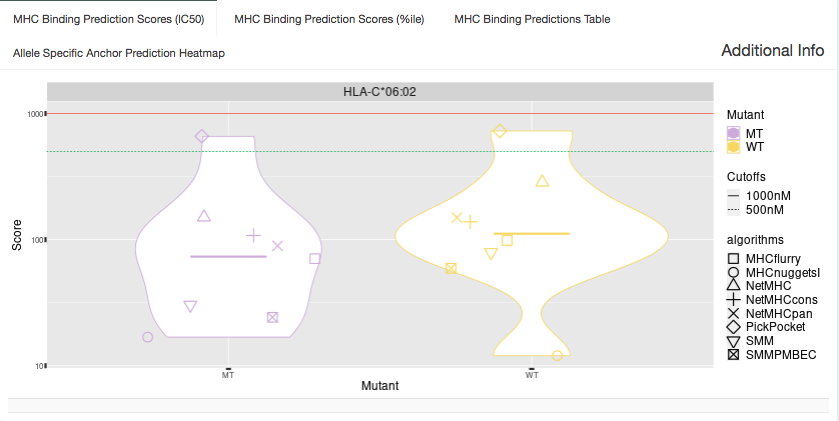
IC50 Plot:
Shown in this tab are violin plots of the individual IC50-based binding affinity predictions of the MT and WT peptides for HLA alleles were the MT binds well to. These peptides each have up to 8 binding algorithm scores for Class I alleles or up to 4 algorithm scores for Class II alleles.
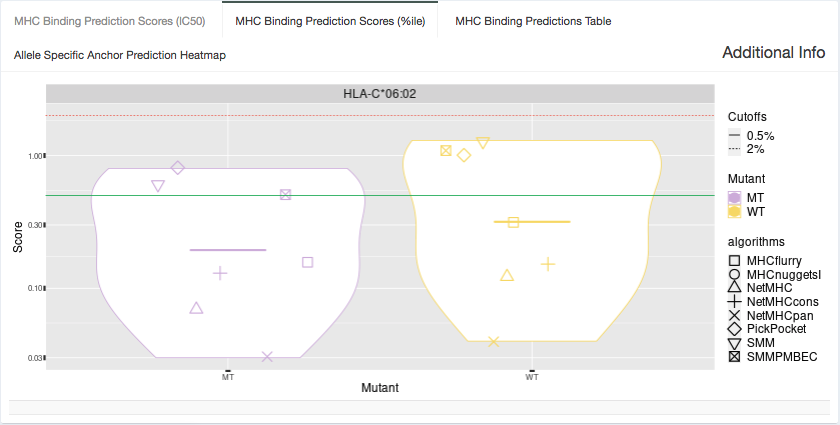
%ile Plot:
Shown in this tab are violin plots of the individual percentile-based binding affinity predictions of the MT and WT peptides for HLA alleles were the MT binds well to. These peptides each have up to 8 binding algorithm scores for Class I alleles or up to 4 algorithm scores for Class II alleles.
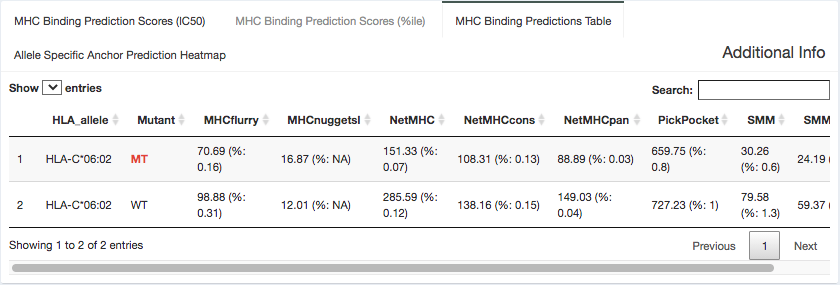
Binding Data:
Here, we provide the specific IC50 and percentile binding affinity predictions generated from each individual algorithm. This data is specific to the MT/WT peptide pair selected in the peptide table.
Elution Data:
Here, we provide the specific elution scores and percentiled generated from each individual algorithm. This data is specific to the MT/WT peptide pair selected in the peptide table.

Allele-specific anchor prediction heatmap:
Previously, our lab has computationally predicted anchor positions for different hla alleles and peptide length combinations (“Accurate neoantigen prediction depends on mutation position relative to patient allele-specific MHC anchor location”). These predictions are normalized probabilities representing the likelihood of each position of the peptide to participate in anchoring to the hla allele. Top 30 MT/WT peptide pairs from the peptide table are shown in this tab with anchor probabilities overlaying as a heatmap. These anchor probabilities shown are both allele and peptide length specific. The mutated amino acid(s) is/are marked in red (or the flanking ones are, in the case of deletions) and each MT/WT pair are separated from others using a dotted line.
