Comparison Tool¶
pVACtools includes a file comparison utility designed to help users compare results across different runs of the same dataset. This tool is particularly useful for identifying changes introduced by updates to pVACtools, such as algorithm improvements or altered default parameters. By highlighting differences between result files, it enables users to validate consistency, track the impact of software changes, and ensure reproducibility in their workflows.
The comparison includes the following pVACseq output files, along with the inputs.yml file,
which records the inputs used for each specific pipeline run:
File Name |
Description |
|---|---|
|
A list of all predicted epitopes and their binding affinity scores, with
additional variant information from the |
|
An aggregated version of the |
|
A file outlining details of reference proteome matches |
|
A JSON file with detailed information about the predicted epitopes, formatted for pVACview. This file, in combination with the aggregated.tsv file, is required to visualize your results in pVACview. Not generated when running only with presentation and immunogenicity algorithms. |
Usage¶
usage: pvactools compare [-h] [--output-dir OUTPUT_DIR] [--mhc-class {1,2}]
[--no-server] [--start-server]
[--aggregated-columns AGGREGATED_COLUMNS]
[--unaggregated-columns UNAGGREGATED_COLUMNS]
[--reference-match-columns REFERENCE_MATCH_COLUMNS]
[results_folder1] [results_folder2]
Run a comparison between two output results folders
positional arguments:
results_folder1 Path to first results input folder (default: None)
results_folder2 Path to second results input folder (default: None)
optional arguments:
-h, --help show this help message and exit
--output-dir OUTPUT_DIR
Specify where the output directory should be generated
(default: compare_output)
--mhc-class {1,2} Specify the MHC class to run: '1' for Class I or '2'
for Class II. If not specified, both classes will be
processed. (default: None)
--no-server If specified, will not start the report server after
the comparisons finish (default: False)
--start-server If specified, will only start the report server and
will not run a comparison (default: False)
--aggregated-columns AGGREGATED_COLUMNS
Comma-separated columns to include in the aggregated
TSV comparison, choices: Gene, AA Change, Num Passing
Transcripts, Best Peptide, Best Transcript, Num
Passing Peptides, IC50 MT, IC50 WT, %ile MT, %ile WT,
RNA Expr, RNA VAF, DNA VAF, Tier, Ref Match (default:
['Num Passing Transcripts', 'Best Peptide', 'Best
Transcript', 'Num Passing Peptides', 'Tier'])
--unaggregated-columns UNAGGREGATED_COLUMNS
Comma-separated columns to include in the unaggregated
TSV comparison, choices: Biotype, Median MT IC50
Score, Median WT IC50 Score, Median MT Percentile,
Median WT Percentile, WT Epitope Seq, Tumor DNA VAF,
Tumor RNA Depth, Tumor RNA VAF, Gene Expression,
BigMHC_EL WT Score, BigMHC_EL MT Score, BigMHC_IM WT
Score, BigMHC_IM MT Score, MHCflurryEL Processing WT
Score, MHCflurryEL Processing MT Score, MHCflurryEL
Presentation WT Score, MHCflurryEL Presentation MT
Score, MHCflurryEL Presentation WT Percentile,
MHCflurryEL Presentation MT Percentile, MHCflurry WT
IC50 Score, MHCflurry MT IC50 Score, MHCflurry WT
Percentile, MHCflurry MT Percentile, MHCnuggetsI WT
IC50 Score, MHCnuggetsI MT IC50 Score, MHCnuggetsI WT
Percentile, MHCnuggetsI MT Percentile, NetMHC WT IC50
Score, NetMHC MT IC50 Score, NetMHC WT Percentile,
NetMHC MT Percentile, NetMHCcons WT IC50 Score,
NetMHCcons MT IC50 Score, NetMHCcons WT Percentile,
NetMHCcons MT Percentile, NetMHCpan WT IC50 Score,
NetMHCpan MT IC50 Score, NetMHCpan WT Percentile,
NetMHCpan MT Percentile, NetMHCpanEL WT Score,
NetMHCpanEL MT Score, NetMHCpanEL WT Percentile,
NetMHCpanEL MT Percentile, PickPocket WT IC50 Score,
PickPocket MT IC50 Score, PickPocket WT Percentile,
PickPocket MT Percentile, SMM WT IC50 Score, SMM MT
IC50 Score, SMM WT Percentile, SMM MT Percentile,
SMMPMBEC WT IC50 Score, SMMPMBEC MT IC50 Score,
SMMPMBEC WT Percentile, SMMPMBEC MT Percentile,
DeepImmuno WT Score, DeepImmuno MT Score, Problematic
Positions, Gene of Interest (default: ['Biotype',
'Median MT IC50 Score', 'Median WT IC50 Score',
'Median MT Percentile', 'Median WT Percentile', 'WT
Epitope Seq', 'Tumor DNA VAF', 'Tumor RNA Depth',
'Tumor RNA VAF', 'Gene Expression'])
--reference-match-columns REFERENCE_MATCH_COLUMNS
Comma-separated columns to include in the reference
match TSV comparison, choices: Peptide, Hit
Definition, Match Window, Match Sequence (default:
['Peptide', 'Match Window'])
Viewing results¶
Once the comparison has completed, a local web server will automatically be launched, provided the --no-server
option was not used. If you wish to start the server manually at a later time, you can do so by running the
comparison tool again with the --start-server parameter.
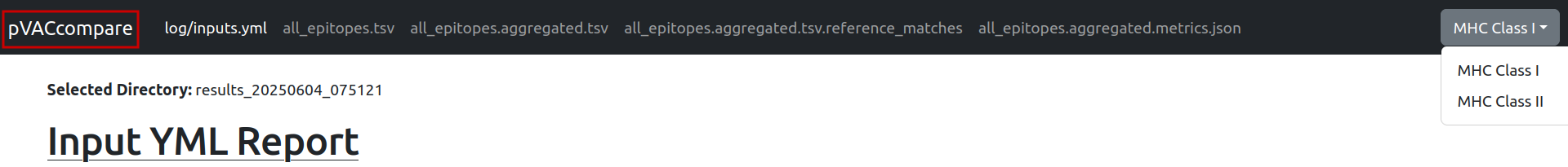
After the server is running, navigate to the provided link in your browser. This will open the report interface, which should resemble the following:
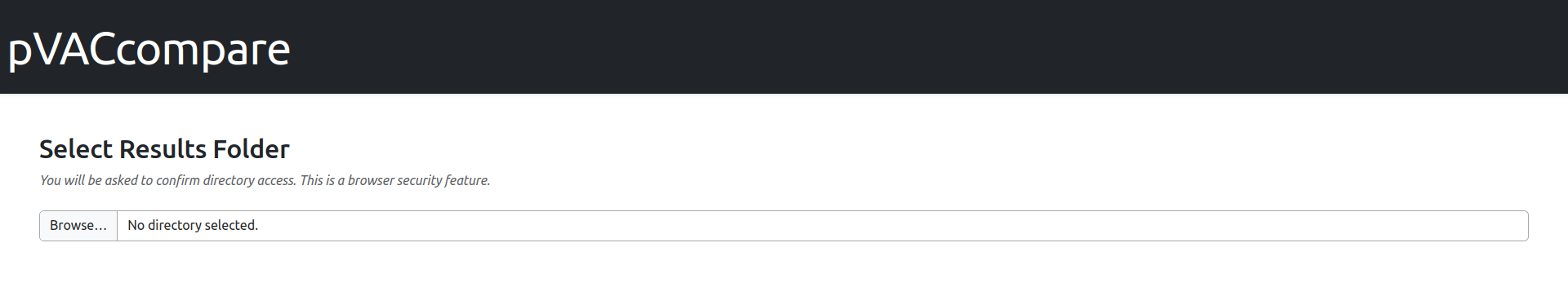
To begin viewing results select the target results folder using the file selection tool, then click the “Confirm and Load Files” button.
This will open a detailed comparison page, where you can review differences across each of the included output files. Use the “pVACcompare” button in the top-left corner to return to the result selection page at any time. Use the MHC class dropdown in the top-right corner to toggle between Class I and Class II results (this option will only be available if results for both classes were included in the comparison).