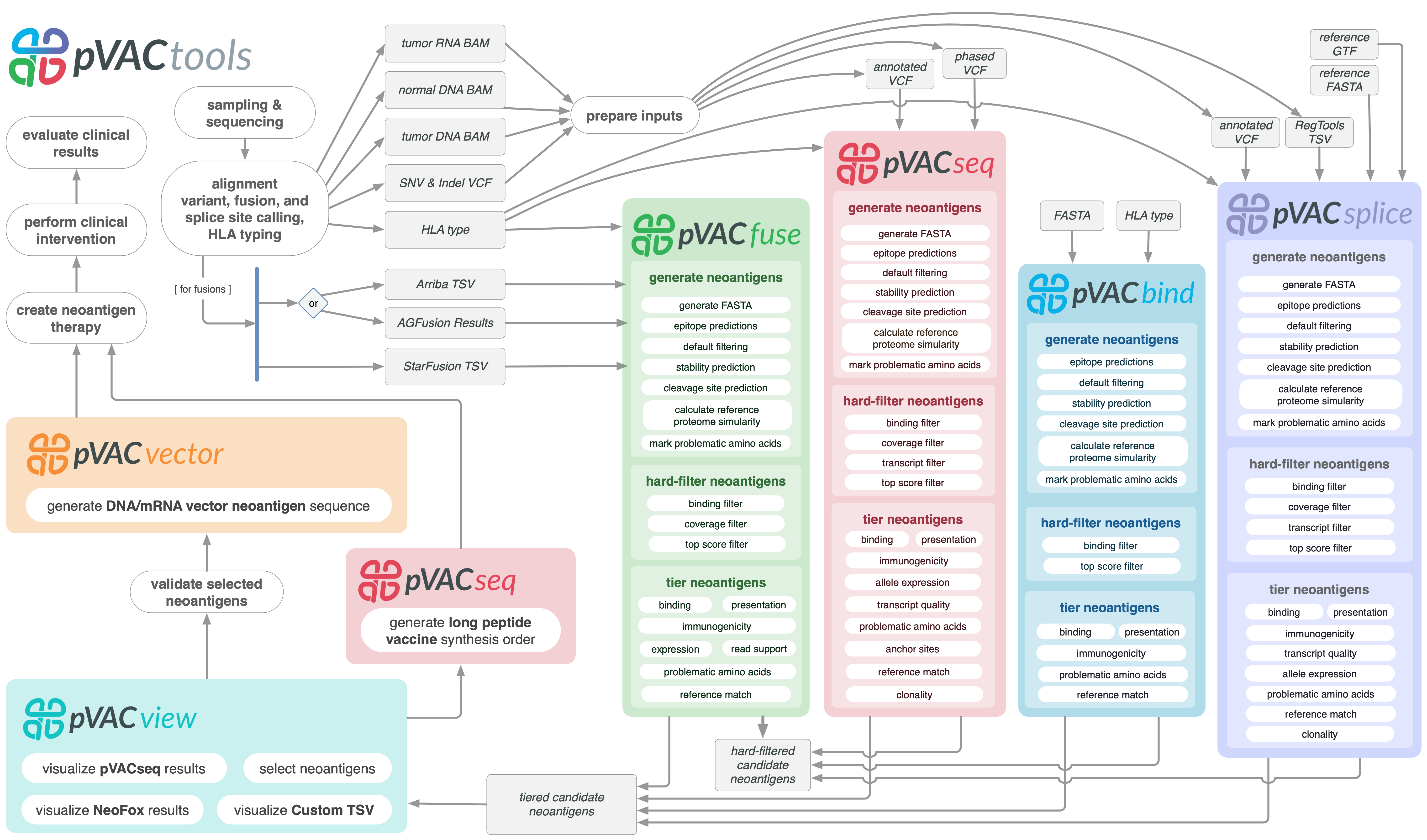
pVACtools¶
pVACtools is a cancer immunotherapy tools suite consisting of the following tools:
- pVACseq
A cancer immunotherapy pipeline for identifying and prioritizing neoantigens from a VCF file.
- pVACbind
A cancer immunotherapy pipeline for identifying and prioritizing neoantigens from a FASTA file.
- pVACfuse
A tool for detecting neoantigens resulting from gene fusions.
- pVACsplice
A tool for detecting neoantigens resulting from splice site variants.
- pVACvector
A tool designed to aid specifically in the construction of DNA-based cancer vaccines.
- pVACview
An application based on R Shiny that assists users in reviewing, exploring and prioritizing neoantigens from the results of pVACtools processes for personalized cancer vaccine design.
Contents¶
Version 7.1.0¶
This is a minor version release. It adds the following features:
pVACtools now supports additional prediction algorithms:
TLBind (binding and presentation)
TLImm (immunogenicity)
These algorithms also support normalized percentiles using the
--use-normalized-percentilesflags. by @susannasiebert in https://github.com/griffithlab/pVACtools/pull/1377
Additionally, it fixes the following problem(s):
Fix tiers argument type to have the most up-to-date tier list by @susannasiebert in https://github.com/griffithlab/pVACtools/pull/1423
Fix NetChop bug that would include position before epitope (0) by @susannasiebert in https://github.com/griffithlab/pVACtools/pull/1432
Fix bug in lowexpr logic by @susannasiebert in https://github.com/griffithlab/pVACtools/pull/1433
Fix tbi file existence check in pVACsplice runs by @susannasiebert in https://github.com/griffithlab/pVACtools/pull/1434
New in Version 7¶
This is a major version release. Please note that pVACtools 7.0 is not guaranteed to be backwards-compatible and certain changes could break old workflows.
New Tools¶
pVACseq now has the option of running machine learning (ML)-based neoantigen prioritization predictions. The ML predictor uses a trained random forest model to predict whether neoantigen candidates in the aggregated report should be evaluated as “Accept”, “Reject”, or “Pending” based on a comprehensive set of features derived from binding affinity predictions, expression data, and variant characteristics. The ML predictor can be enabled by adding the
--run-ml-predictionsparameter to a pVACseq run. More details can be found in the output file documentation, the vignette.
New Features¶
pVACtools now supports multiple additional prediction algorithms:
MixMHCpred (class I binding score and percentile)
MixMHC2pred (class II binding score and percentile)
PRIME (class I immunogenicity score and percentile)
ImmuScope (class II immunogenicity score)
In order to support a more comprehensive evaluation of candidates, aggregate binding, presentation, and immunogenicity information is now available in the final reports and is used to filter, prioritize, and tier candidates:
Best and median binding percentiles, presentation percentiles and immunogenicity percentiles are now calculate in the all_epitopes.tsv, filtered.tsv, and aggregated.tsv files in addition to the previously available combined percentiles that were aggregating percentile ranks over all prediction algorithms regardless of algorithm type.
Three new parameters -
--binding-percentile-threshold,--presentation-percentile-threshold, and--immunogenicity-percentile-thresholdreplace the old--percentile-threshold. These three new thresholds have been updated to use a default of 2.0 instead of not having a default. This means that filtering and tiering will now by default include evaluation of binding, presentation, and immunogenicity percentiles.The aggregate report
PoorBindingtier now evaluates the IC50 binding affinity as well as the binding percentile. Candidates failing either threshold (when aconservative--percentile-threshold strategyis selected, default) or both thresholds (when aexploratory--percentile-threshold-strategyis selected) will be binned in this tier when all other evaluation criteria are passed.Two new tiers,
PoorPresentationandPoorImmunogenicity, are added to bin candidates that failed the--presentation-percentile-thresholdor--immunogenicity-percentile--threshold, respectively, when all other evaluation criteria are passed.pVACvector has been updated to work on the binding percentile instead of the combined percentile. The corresponding parameter has been renamed to
--binding-percentile-thresholdwith a new default of 2.0. The junction output file header recording each junction’s binding percentile has been updated frompercentiletobinding_percentileto reflect this change.pVACview has been updated to display more information regarding immunogenicity and presentation scores.
pVACtools runs that do not use a binding predictor would previously skip the binding filter, top score filter, and the aggregate report creation. These steps will now be run.
The
--top-score-metric2has been updated for sorting candidates and determining the criteria for selecting the Best Peptide (in the aggregate report) and top candidate (in the top score filter). It is now a list of criteria to consider. All listed criteria are assigned a rank and the sum of those ranks is used. By default both the IC50 (ic50) and the combined percentile (combined_percentile) are used. Other allowed values are the binding percentile (binding_percentile), the presentation percentile (presentation percentile), and the immunogenicity percentile (immunogenicity_percentile). Any number and combination of these five criteria may be specified.Not all prediction algorithms supported by pVACtools may support a percentile rank. In order to alleviate this issue, and to provide percentile ranks that have been consistently calculated, we have run predictions for all class I algorithms supported by pVACtools on 100,000 reference peptides each in lengths 8-11 and for the most common 1,000 human class I MHC alleles. These predictions support a new feature in pVACtools: normalized percentiles (
--use-normalized-percentiles). With this option enabled, any of the pVACtools pipelines will calculate normalized percentiles scores for all predicted neoantigen candidates and selected prediction algorithms. These normalized percentile ranks will be used in place of percentile ranks calculated by the algorithms natively. Predictions for allele or lengths we have not calculated reference scores will result inNApercentile ranks. Turning on this option in class II runs or with non-human data will be ignored. The peptides used in our predictions and the raw scores we calculated are available at https://github.com/griffithlab/pvactools_percentiles_data.In pVACbind and pVACfuse the
Mutationcolumn name in the various report files has been renamed toIndexin order to ensure consistency between these and other tools.
Past release notes can be found on our Release Notes page.
To stay up-to-date on the latest pVACtools releases please join our Mailing List.
Citations¶
Jasreet Hundal , Susanna Kiwala , Joshua McMichael, Chris Miller, Huiming Xia, Alex Wollam, Conner Liu, Sidi Zhao, Yang-Yang Feng, Aaron Graubert, Amber Wollam, Jonas Neichin, Megan Neveau, Jason Walker, William Gillanders, Elaine Mardis, Obi Griffith, Malachi Griffith. pVACtools: A Computational Toolkit to Identify and Visualize Cancer Neoantigens. Cancer Immunology Research. 2020 Mar;8(3):409-420. doi: 10.1158/2326-6066.CIR-19-0401. PMID: 31907209.
Jasreet Hundal, Susanna Kiwala, Yang-Yang Feng, Connor J. Liu, Ramaswamy Govindan, William C. Chapman, Ravindra Uppaluri, S. Joshua Swamidass, Obi L. Griffith, Elaine R. Mardis, and Malachi Griffith. Accounting for proximal variants improves neoantigen prediction. Nature Genetics. 2018, DOI: 10.1038/s41588-018-0283-9. PMID: 30510237.
Jasreet Hundal, Beatriz M. Carreno, Allegra A. Petti, Gerald P. Linette, Obi L. Griffith, Elaine R. Mardis, and Malachi Griffith. pVACseq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Medicine. 2016, 8:11, DOI: 10.1186/s13073-016-0264-5. PMID: 26825632.
Huiming Xia, My H. Hoang, Evelyn Schmidt, Susanna Kiwala, Joshua McMichael, Zachary L. Skidmore, Bryan Fisk, Jonathan J. Song, Jasreet Hundal, Thomas Mooney, Jason R. Walker, S. Peter Goedegebuure, Christopher A. Miller, William E. Gillanders, Obi L. Griffith, Malachi Griffith. pVACview: an interactive visualization tool for efficient neoantigen prioritization and selection. Genome Medicine. 2024, 16:132, DOI: 10.1186/s13073-024-01384-7. PMID: 39538339.
Source code¶
The pVACtools source code is available in GitHub.
License¶
This project is licensed under BSD 3-Clause Clear License.